Fungal Cell Structure and Targets
Knowledge of fungal cell structure and function is essential for understanding the pharmacology of antifungal agents. Like mammalian cells, fungi are eukaryotes with DNA organized into chromosomes within the cell nucleus and have distinct cytoplasmic organelles including endoplasmic reticulum, Golgi apparatus, mitochondria, and storage vacuoles. This homology to mammalian cells also extends to biosynthetic pathways, where fungi share similar mechanisms for DNA replication and protein synthesis.
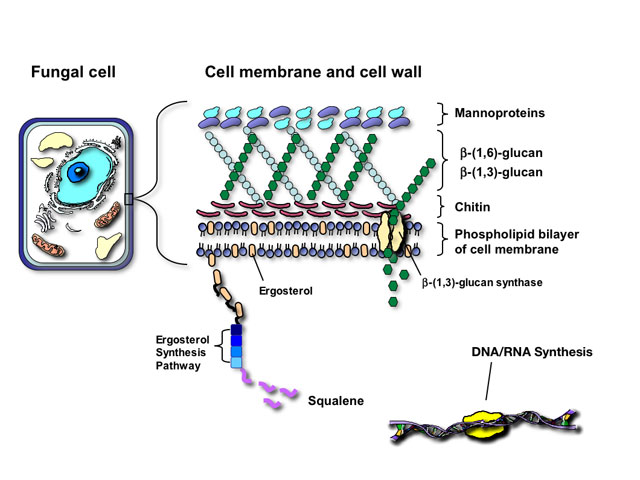
The similarity of fungal and mammalian cells creates a number of problems for designing drugs that are selectively toxic to fungal cells but not the human host. Antifungal agents currently utilized for mycoses attack one of four targets in fungal pathogens:
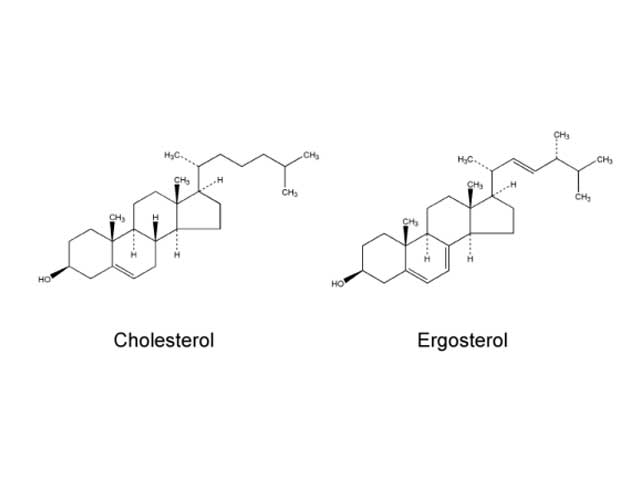
Fungi and mammalian cells both contain a cell membrane that serves and important role in cell structure, division, and metabolism. Complex lipid particles, called sterols, account for approximately 25% of the weight of the cell membrane. However, the sterol content between mammalian cells and fungal cells is different. Whereas mammalian cell membranes contain primarily cholesterol, ergosterol is the predominant sterol in many pathogenic fungi. This difference in sterol content has been exploited as the target of antifungal drug action by several classes of antifungal agents currently used to treat superficial and invasive fungal infections including the polyenes, azoles, and allylamines.
Polyene antifungals
Polyene antifungals such as amphotericin B act by binding to ergosterol in the fungal cell membrane. This binding results in depolarization of the membrane and formation of pores that increase permeability to proteins and monovalent and divalent cations, eventually leading to cell death. Amphotericin B may also induce oxidative damage in fungal cells and has been reported to stimulate of host immune cells.
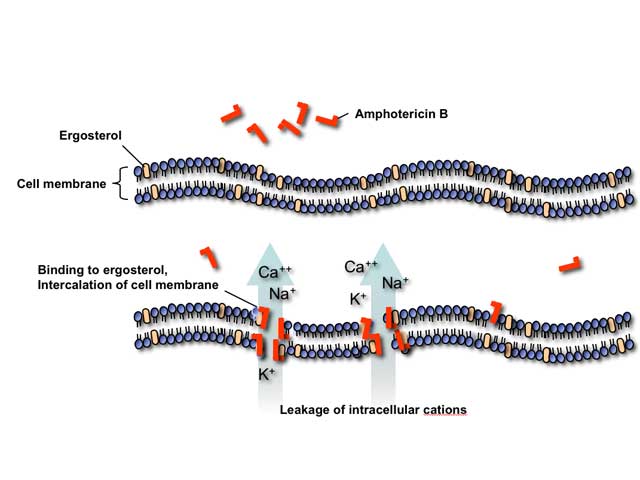
Toxicities of polyene antifungals are an extension of their mechanism of action. Stimulation of the host immune cells by amphotericin B causes release of inflammatory cytokines by circulating monocytes resulting in fever, chills, rigor, nausea, vomiting, myalgias, arthralgias, and headache during intravenous infusions. At higher concentrations, amphotericin B binds to cholesterol in mammalian cell membranes leading to various organ toxicities, most importantly nephrotoxicity.
Azole antifungals
Azole antifungals inhibit the fungal cytochrome P-450 3-A dependent enzyme 14-alpha demethylase, thereby interrupting the synthesis of ergosterol. Inhibition of this critical enzyme in the ergosterol synthesis pathway leads to the depletion of ergosterol in the cell membrane and accumulation of toxic intermediate sterols, causing increased membrane permeability and inhibition of fungal growth.
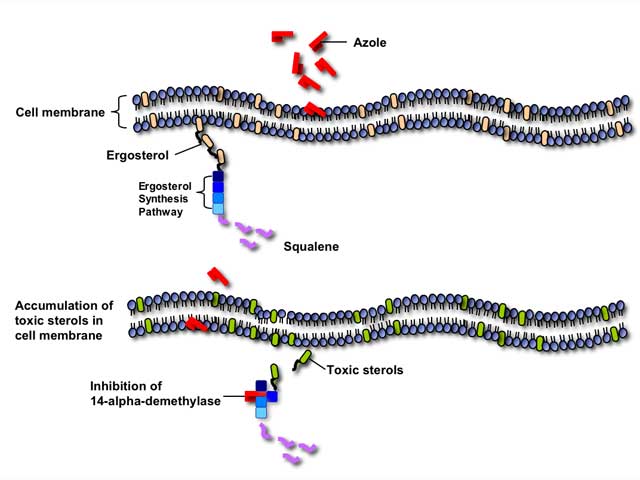
Azole antifungals can also inhibit many mammalian cytochrome P450-dependent enzymes involved in hormone synthesis or drug metabolism. Therefore, azole antifungals are particularly susceptible to clinically-significant drug interactions with other medications metabolized through the P450 pathway.
Allylamines
Allylamines work in a conceptually similar fashion to azole antifungals by inhibiting the synthesis of ergosterol. However, allylamines act at an earlier step in the ergosterol synthesis pathway by inhibiting the enzyme squalene epoxidase. Like the azoles, terbinafine has the potential for drug interactions with other medications metabolized through the mammalian cytochrome P-450 pathway.
The fungal cell wall is critical for cell viability and pathogenicity. Beyond serving as a protective shell and providing cell morphology, the fungal cell wall is a critical site for exchange and filtration of ions and proteins, as well as metabolism and catabolism of complex nutrients. Because mammalian cells lack a cell wall, it also represents an ideal and specific target for antifungal therapy.
Structurally, the fungal cell wall is composed of a complex network of proteins and polycarbohydrates that varies in composition depending on the fungal species. Disruption of this protein/carbohydrate matrix results in a structurally-defective cell wall, rendering the fungal cell sensitive to osmotic lysis.
Glucan synthesis inhibitors
The glucan synthesis inhibitors are, collectively, agents that are presumed to block fungal cell wall synthesis by inhibiting the enzyme 1,3-beta glucan synthase. Inhibition of this enzyme results in depletion of glucan polymers in the fungal cell, resulting in an abnormally weak cell wall unable to withstand osmotic stress.
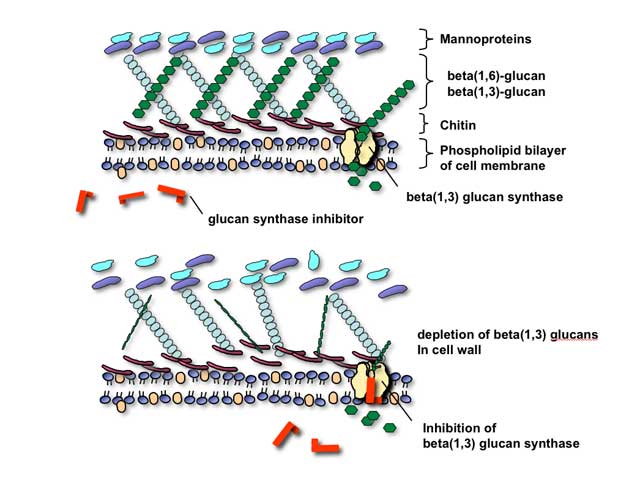
As technical issues in the laboratory have made formal proof of the assertion that this enzyme is actual target of these compounds, it is most correct to speak of them at present as being glucan synthesis inhibitors rather than glucan synthase inhibitors. There are three such agents at present, with all three belonging to the chemical family also known as the echinocandins: caspofungin, micafungin, and anidulafungin. As would be predicted from their mechanism of action, these agents appear to be well-tolerated and have relatively fewer toxicities than polyene or azole-class antifungals. Indeed, maximum tolerated dosages were not reached, in human volunteers, for these agents in phase II clinical trials.
DNA/RNA Synthesis:
DNA and protein synthesis have historically been difficult targets for the development of selectively-toxic antifungal therapy, as fungal and mammalian cells share remarkable homology in DNA replication and RNA translation. However, advances in molecular biology and functional genomics are beginning to highlight important differences between mammalian and fungal cells that could be exploited for the development of new antifungal therapies. For the time being, only one class of agents in clinical use targets DNA/RNA synthesis.
Antimetabolites
This class has only one example, flucytosine or 5-fluorocytosine (5-FC). Flucytosine was originally developed in the 1950’s as a potential antineoplastic agent. Although ineffective against tumors it was later found to have antifungal activity. This small molecule is transported into susceptible fungal cells by a specific enzyme cytosine permease and converted in the cytoplasm by cytosine deaminase to 5-fluorouracil (5-FU)- a pyrimidine anti-metabolite used as chemotherapy for many types of colorectal cancer.
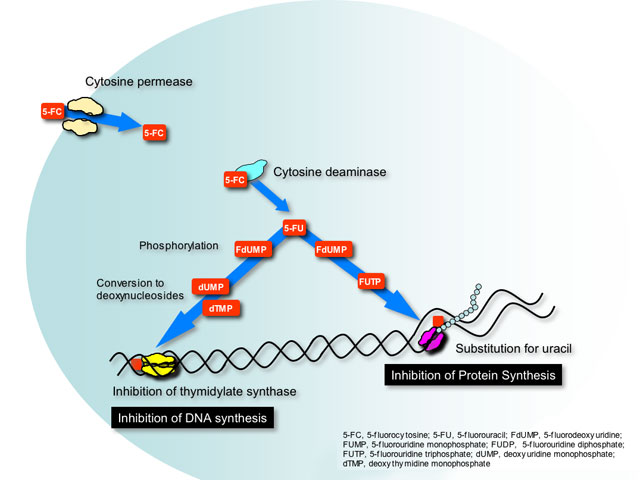
5-FU is phosphorylated and incorporated into RNA where it causes miscoding and halts protein synthesis. Additionally, phosphorylated 5-FU is converted to its deoxynucleoside, which inhibits DNA synthesis by blocking the functions of a key enzyme in DNA replication- thymidylate synthetase.
Flucytosine can be converted to 5-FU by bacteria residing in the gastrointenstinal tract. Not surprisingly, the most common adverse effects seen with flucytosine are similar to 5-FU chemotherapy (diarrhea, nausea and vomiting, bone marrow suppression) but at reduced intensity.
Miscellaneous Agents:
This class contains only one agent, griseofulvin. Griseofulvin inhibits fungal cell mitosis by disrupting mitotic spindle formation-a critical step in cellular division.